Estimating molecular volumes to aid in powder X-ray diffraction indexing
An overview of using database-derived atomic volumes to aid PXRD indexing.
PXRD
Indexing
Author
Mark Spillman
Published
November 10, 2021
Introduction
An article published in 2001 by D. W. M. Hofmann describes how crystallographic databases can be used to derive the average volume occupied by atoms of each element in crystal structures. Using his tabulated values, it’s possible to rapidly estimate the volume occupied by a given molecule, and use this to aid indexing of powder diffraction data. This is particularly useful for laboratory diffraction data, which is generally associated with lower figures of merit such as de Wolff’s \(M_{20}\) and Smith and Snyder’s \(F_N\), which can make discriminating between alternative options more challenging. Other volume estimation methods, notably the 18 ų rule are also commonly used, though Hofmann’s volumes give generally more accurate results.
I’ve put together a freely available web-app, HofCalc, which can be used to conveniently obtain these estimates. It should display reasonably well on mobile devices as well as PCs/laptops. You can access it at the following address:
This post will explain how it works, and will look at some examples of how it can be used in practice. I’m grateful to Norman Shankland who provided invaluable feedback and assistance with debugging of the app.
Hofmann volumes
After applying various filters to crystal structures deposited in the CSD, Hofmann ended up with a dataset comprised of 182239 structures. Hofmann only considers the elements up to atomic number 100 (fermium) in his work, and assumes that the volume of the unit cell is equivalent to:
Where \(n_i\) is the number of atoms of element \(i\) in the unit cell, and \(\bar{v_i}\) is the average volume occupied by an atom of element \(i\). He also assumes that atomic volumes vary linearly with temperature.
He split the dataset into 20 subsets, then used an iterative least-squares method to solve the above equation for each of the subsets. This allowed him to find the average volumes occupied by atoms of each element, and due to the splitting of the data into subsets, he also obtains their standard deviations. The coefficient of thermal expansion, \(\bar{\alpha}\), was found to be \(0.95 \times 10^{-4} K^{-1}\). This temperature correction factor then allowed him to provide the average volumes for all of the elements represented in the CSD at 298 K.
You can download a .json file containing the 298 K volumes here.
Comparison with other atomic volumes
Let’s compare Hofmann’s volumes to those obtained from other sources. Hofmann’s article compares his volumes to those derived by Mighell and coworkers, which were published in 1987. As additional comparison points, which highlight the importance of using crystallographic volumes in this context, I also downloaded some atomic radii data from Wikipedia and converted these into atomic volumes (assuming spherical atoms). Sources for these radii may be found at the bottom of the Wikipedia article.
Click on the coloured boxes in the top left to view individual types of volume, and shift-click to add other volumes back in again.
Code
import jsonimport pandas as pdimport numpy as npimport altair as altwithopen("files/Hofmann-volumes.json") as hv: hofmann_volumes = json.load(hv)hv.close()vols = []for i, key inenumerate(hofmann_volumes.keys()): vols.append([i+1, key, hofmann_volumes[key]])df = pd.DataFrame(vols)df.columns = ["Atomic number", "Element", "Hofmann"]df.reset_index(drop=True, inplace=True)df.replace("N/A", np.NaN, inplace=True)wikiradii = pd.read_excel("files/wikipedia_radii.xlsx")wikiradii.replace("", np.NaN, inplace=True)radtype = ["Mighell", "Empirical","Calculated","vdW","Covalent-single","Covalent-triple","Metallic"]for r in radtype:# Radii are in pm so /100 to convert to angstroms.if r =="Mighell": df[r] = wikiradii[r].values.astype(float)else: df[r] = (4*np.pi/3)*(wikiradii[r].values.astype(float)/100)**3# Convert our dataframe to long-form as this is what is expected by altairdflong = df.melt("Atomic number", var_name="Volume", value_vars=["Hofmann"] + radtype)# Restore the element symbols to the long dataframeelement = []for an in dflong["Atomic number"]: element.append(df["Element"][df["Atomic number"] == an].item())dflong["Element"] = elementclick = alt.selection_multi(encodings=["color"])# scatter plot, modify opacity based on selectionscatter = alt.Chart(dflong).mark_point().encode( x=alt.X('Element:N',sort=dflong["Atomic number"].values), y=alt.Y("value:Q", axis=alt.Axis(title='Volume / ų')), tooltip=['Element', 'Volume:N', 'value'], opacity=alt.value(0.85), color="Volume:N").transform_filter(click).properties(width=650, height=500).interactive()# legendlegend = alt.Chart(dflong).mark_rect().encode( y=alt.Y('Volume:N', axis=alt.Axis(title='Select volume'), sort=[4,6,0,1,2,3,5,7]), color=alt.condition(click, 'Volume:N', alt.value('lightgray'), legend=None),).properties( selection=click,)chart = (legend | scatter)chart
As you can see, the volumes of Hofmann and Mighell differ significantly from those I derived from the atomic radii.
Let’s print out some of the statistics describing the data, as well compare the coefficient of variation for each type of volume.
Code
df.describe()[["Hofmann"]+radtype]
Hofmann
Mighell
Empirical
Calculated
vdW
Covalent-single
Covalent-triple
Metallic
count
84.000000
100.00000
91.000000
86.000000
55.000000
95.000000
71.000000
68.000000
mean
39.586310
42.65840
17.084519
23.849111
37.865054
13.250696
6.963913
19.946436
std
13.460829
17.12722
12.248898
20.652779
33.870162
8.900926
3.301590
12.541749
min
5.080000
5.40000
0.065450
0.124788
7.238229
0.137258
0.623615
5.884949
25%
31.000000
30.87500
10.305995
7.377367
19.870146
7.329463
4.988916
11.039115
50%
39.300000
41.00000
12.770051
19.334951
27.833137
11.008442
6.538266
17.157285
75%
49.250000
53.50000
23.632685
36.484963
39.070796
19.160766
9.204406
24.469830
max
74.000000
85.00000
77.951815
110.850435
176.533179
52.306127
16.837592
77.951815
Code
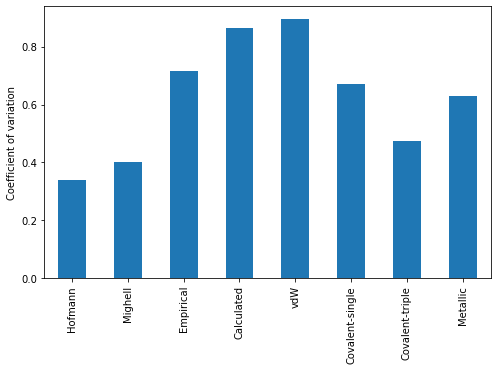
import matplotlib.pyplot as pltplt.figure(figsize=(8,5))((df.describe().loc["std"] / df.describe().loc["mean"])[["Hofmann"]+radtype]).plot.bar()plt.ylabel("Coefficient of variation")plt.show()
We see a much lower coefficient of variation for Hofmann’s volumes than the others.
HofCalc - using the web app
HofCalc makes use of two key python libraries to process chemical formulae (pyvalem) and resolve chemical names (PubChemPy) prior to processing. This allows the app to have a really convenient interface for specifying queries (see below), which enables users to easily mix and match between formulae and names to obtain the information they need.
Formulae and names
Basic use
The simplest option is to enter the chemical formula or name of the material of interest. Names are resolved by querying PubChem, so common abbreviations for solvents can often be used e.g. DMF. Note that formulae can be prefixed with a multiple, e.g. 2H2O
Search term
Type
\(V_{Hofmann}\)
ethanol
name
69.61
CH3CH2OH
formula
69.61
water
name
21.55
2H2O
formula
43.10
Multiple search terms
It is also possible to search for multiple items simultaneously, and mix and match name and formulae by separating individual components with a semicolon. This means that for example, ‘amodiaquine dihydrochloride dihydrate’ can also be entered as ‘amodiaquine; 2HCl; 2H2O’.
Search term
Total \(V_{Hofmann}\)
carbamazepine; L-glutamic acid
497.98
zopiclone; 2H2O
496.02
C15H12N2O; CH3CH2COO-; Na+
419.79
sodium salicylate; water
204.21
amodiaquine dihydrochloride dihydrate
566.61
amodiaquine; 2HCl; 2H2O
566.61
More complex examples - hemihydrates
In cases where fractional multiples of search components are required, such as with hemihydrates, care should be taken to check the evaluated chemical formula for consistency with the expected formula.
Search term
Evaluated as
\(V_{Hofmann}\)
Divide by
Expected Volume
Calcium sulfate hemihydrate
Ca2 H2 O9 S2
253.07
2
126.53
calcium; calcium; sulfate; sulfate; water
Ca2 H2 O9 S2
253.07
2
126.53
calcium; sulfate; 0.5H2O
Ca1 H1.0 O4.5 S1
126.53
-
126.53
Codeine phosphate hemihydrate
C36 H50 N2 O15 P2
1006.77
2
503.38
codeine; codeine; phosphoric acid; phosphoric acid; water
C36 H50 N2 O15 P2
1006.77
2
503.38
codeine; phosphoric acid; 0.5H2O
C18 H25.0 N1 O7.5 P1
503.38
-
503.38
Charged species in formulae
Charges could potentially interfere with the parsing of chemical formulae. For example, two ways of representing an oxide ion:
Search term
Evaluated as
O-2
1 x O
O2-
2 x O
Whilst is is recommended that charges be omitted from HofCalc queries, if including charges in your queries, ensure that the correct number of atoms has been determined in the displayed atom counts or the downloadable summary file. For more information on formatting formulae, see the pyvalem documentation.
Temperature
The temperature, \(T\) (in kelvin) is automatically included in the volume calculation via the following equation:
\[V = \sum{n_{i}v_{i}}(1 + \alpha(T - 298))\]
Where \(n_{i}\) and \(v_{i}\) are the number and Hofmann volume (at 298 K) of the \(i\)th element in the chemical formula, and \(\alpha = 0.95 \times 10^{-4} K^{-1}\).
Unit cell volume
If the volume of a unit cell is supplied, then the unit cell volume divided by the estimated molecular volume will also be shown.
Search term
\(V_{cell}\)
\(V_{Hofmann}\)
\(\frac{V_{cell}}{V_{Hofmann}}\)
zopiclone, 2H2O
1874.61
496.02
3.78
verapamil, HCl
1382.06
667.57
2.07
Summary Files
Each time HofCalc is used, a downloadable summary file is produced. It is designed to serve both as a record of the query for future reference and also as a method to sense-check the interpretation of the entered terms, with links to the PubChem entries where relevant. An example of the contents of the summary file for the following search terms is given below.
The crystal structure of chlorothiazide N,N-dimethylformamide, a.k.a CT-DMF2, was solved from laboratory powder diffraction data back in 2007. I decided to try re-indexing the diffraction data to see if HofCalc would be of use.
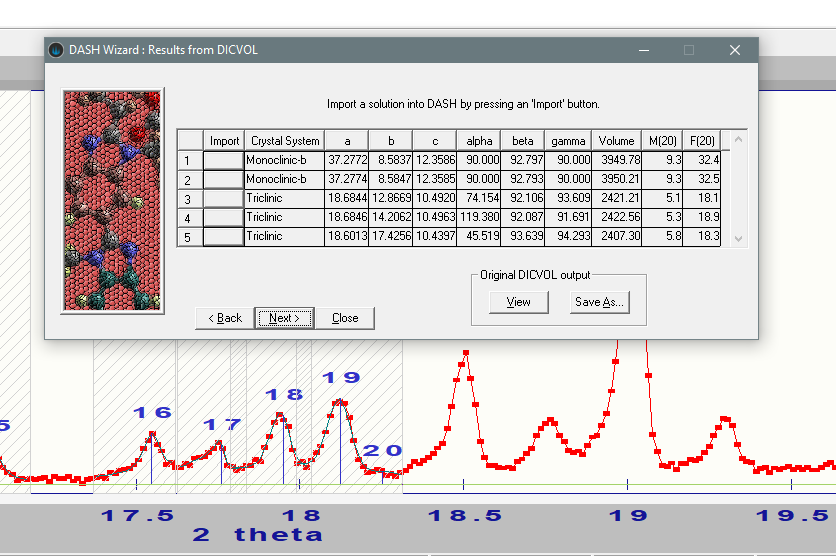
Using the DASH interface to DICVOL, the following unit cells are suggested:
Both monoclinic and triclinic cells are obtained with very different unit cell volumes. Whilst the figures of merit certainly push towards accepting the conclusion of a monoclinic unit cell, it’s worth checking to see if this makes sense given the expected composition of the material. In addition, there may be more than one dimethylformamide molecule crystallising with the chlorothiazide - HofCalc may be able to shed some light there too.
The paper states that the solvate was formed by recrystallisation of chlorothiazide from DMF solvent, so it seems logical to try the following permutations: 1. chlorothiazide alone 2. chlorothiazide + 1 DMF 3. chlorothiazide + 2 DMF (etc)
HofCalc query
\(V_{Hofmann}\)
\(V_{cell}\)
\(\frac{V_{cell}}{V_{Hofmann}}\)
chlorothiazide
284.73
2422 (triclinic)
8.51
chlorothiazide
284.73
3950 (monoclinic)
13.87
chlorothiazide; DMF
385.09
2422 (triclinic)
6.29
chlorothiazide; DMF
385.09
3950 (monoclinic)
10.26
chlorothiazide; DMF; DMF
485.45
2422 (triclinic)
4.99
chlorothiazide; DMF; DMF
485.45
3950 (monoclinic)
8.14
chlorothiazide; DMF; DMF; DMF
585.81
2422 (triclinic)
4.13
chlorothiazide; DMF; DMF; DMF
585.81
3950 (monoclinic)
6.74
If we exclude those results with \(\frac{V_{cell}}{V_{mol}}\) ratios > 0.25 away from a (crystallographically sensible) whole number, we can see from the table that the most favourable compositions are CT + 2xDMF (monoclinic) and CT + 3xDMF (triclinic). Given the higher figure of merit for the monoclinic unit cell, it seems reasonable to take this forward and attempt space-group determination. Doing this in DASH identifies the most probable space group as \(P2_1/c\), which then implies \(Z'=2\). This is indeed the correct result.
If we compare this to the commonly used 18 ų rule, we end up with the following results:
Possible composition
\(V_{18Å^{3}}\)
\(V_{cell}\)
\(\frac{V_{cell}}{V_{18Å^{3}}}\)
chlorothiazide
306
2422 (triclinic)
7.92
chlorothiazide
306
3950 (monoclinic)
12.91
chlorothiazide; DMF
396
2422 (triclinic)
6.12
chlorothiazide; DMF
396
3950 (monoclinic)
9.97
chlorothiazide; DMF; DMF
486
2422 (triclinic)
4.98
chlorothiazide; DMF; DMF
486
3950 (monoclinic)
8.13
chlorothiazide; DMF; DMF; DMF
576
2422 (triclinic)
4.20
chlorothiazide; DMF; DMF; DMF
576
3950 (monoclinic)
6.86
Again, CT + 2xDMF is in the candidates to check, however, using the 18 ų rule, a triclinic pure chlorothiazide unit cell also becomes a viable possibility. Had there been a less clear distinction in the indexing figure-of-merit, this may have resulted in time being wasted on testing this additional possibility.
Conclusions
Hofmann’s volumes give more accurate estimates of molecular volumes in crystals, and should be used in preference to the 18 ų rule where possible.
To make this easier for people, the HofCalc web-app can be used to very rapidly and conveniently obtain these estimates.