Abbreviations I’ll be using: - SDPD = Crystal structure determination from powder diffraction data - PXRD = Powder X-ray diffraction data - GO = Global optimisation - LO = Local optimisation - GPU = Graphics processing unit - CPU = Central processing unit - SA = Simulated annealing - PSO = Particle swarm optimisation - ML = Machine learning
Background
GALLOP is the culmination of several years of work, which kicked off back in 2010 with an article published by Kenneth Shankland and co-workers, who showed that contrary to the wisdom at the time, local optimisation (LO) algorithms were capable of solving the crystal structures of small molecules, provided that several tens of thousands of attempts from random starting positions on the \(\chi^{2}\) hypersurface were performed. In addition to solving the crystal structure, this also gives the locations of the stationary points on the hypersurface.
Interestingly, they showed that using this method, the global minimum on the hypersurface was located more frequently than any other minimum. This indicates that “the topology of the surface is such that the net ‘catchment’ area of stationary points with very low values of \(\chi^{2}\) is significantly larger than that of the vast majority of stationary points.” The figure below, taken from the article, shows the distribution of \(\chi^{2}\) values for stationary points on the 15-dimensional hypersurface for capsaicin.
Shankland et al, 2010
I carried on investigating this method as part of my PhD, and my results confirmed that this approach is effective at solving crystal structures, even of high-complexity (up to 42 degrees of freedom!). However, despite the intriguing results, the approach was not adopted on a wide scale by the SDPD community, perhaps because the performance it offers is approximately the same existing GO-based programs. The code I was using was written in C++, a language I do not know at all well, so I was unable to contribute much to its further development.
A few years after finishing my PhD, I decided I wanted to try writing my own SDPD code in Python. Whilst Python is notoriously slow, my rationale was that Python is much easier to learn than C++, so should provide a lower barrier to entry for people seeking to try out new ideas for SDPD. My first prototype used numpy to try to speed up the code, and borrowed heavily from pymatgen, a fantastic open-source library with lots of crystallographic functionality. Eventually with some help from Kenneth, I had a system which allowed me to easily try out lots of different algorithms, such as those included in scipy.optimize, which features a variety of local and global optimisation algorithms.
In parallel to this, it seemed like every day incredible new results from the field of deep learning were coming out, showing state-of-the-art performance in wide variety of domains. Most neural networks are trained using backpropagation, an algorithm which makes use of automatic differentiation to calculate the gradient of the cost function (which provides a measure of how well the neural network is performing its task) with respect to the parameters of the neural network. Variants of stochastic gradient descent are then used to modify the parameters of the neural network in order to improve the performance of the neural network as measured by the cost function. Whilst neural networks have been in use for over half a century, part of the reason for the explosion in activity was the availability of GPUs and tools to leverage their parallel processing capabilities.
I took an interest in this, and quickly realised that most of the libraries used for this work had well supported python APIs. Some of them, such as PyTorch, are so similar to numpy that it seemed logical to try to port my code to make use of these libraries. This would give both GPU-acceleration and automatic differentiation capabilities for relatively little effort!
Rationale for GALLOP
With my code now capable of running on GPUs, it might seem obvious to implement GPU-versions of commonly used existing algorithms for SDPD such as simulated annealing (SA), parallel tempering and others. However, despite the parallel processing capabilities of GPUs, I found that the performance with GO methods is not particularly good (at least with my code!). Using SA as an example, then yes, it’s possible to run thousands of simultaneous runs on a single GPU, but the number of iterations that can be performed per second is laughably slow in comparison to performance on a CPU. Therefore, because algorithms like SA take a large number of iterations converge, the performance benefits of parallel processing are offset by the amount of time needed to process the large number of iterations required to reach the global minimum.
In contrast to GO algorithms, LO algorithms with access to gradients converge much more rapidly. The automatic differentiation capabilities provided by PyTorch allow gradients to be calculated rapidly, without any additional code to be written. The gradients so obtained are exactly equivalent to the analytical gradient, and are obtained much more rapidly than the approximate gradient that would be obtained via the method of finite differences. Therefore, when processing large numbers of LO runs on a GPU, because they converge much more rapidly than GO methods, you don’t need to wait for a long time to know if any of the runs have been successful!
The next piece of the puzzle is the idea that even if the global minimum is not located (i.e. the structure hasn’t yet been solved), the previously optimised positions may contain some useful information about the crystal structure. This might not be obvious at first, but let’s try to convince ourselves by taking a look at the hypersurface of verapamil hydrochloride, a structure with 23 degrees of freedom. This interactive figure shows a 2D slice through the hypersurface with all degrees of freedom set to their correct crystallographic values, apart from the fractional coordinates of the verapamil molecule along a and b, which form the axes plotted here.
I’ll write another blog post in the future showing how to use the GALLOP code to generate plots like this.
There are several local minima present, each of which represents an incorrect structure that has either 21 or 22 of its 23 degrees of freedom correctly determined. Despite this, the \(\chi^{2}\) at each local minimum gives no indication that the result is so close to correct. This means that even failed runs with high values of \(\chi^{2}\) may be close to the global minimum. If we could accumulate and exploit this information to influence where the next set of local optimisations start, then it might be possible to save time in searching for the global minimum.
With this idea in mind, I tried a few things to attempt to recycle information from “failed” local optimisation attempts, including using kernel density estimation to try to resample various degrees of freedom depending based on the density of solutions that ended up with particular coordinates. This definitely showed an improvement relative to random sampling, but proved inconsistent in terms of the level of improvement obtained. Perhaps I’ll revisit this in a future blog post as I still think there’s something there that may be of use.
Eventually, I ended up trying particle swarm optimisation (PSO) to attempt to recycle the optimised positions. A few things attracted me to PSO: 1. It’s a simple algorithm to implement - just a few lines of code got it working as a proof of concept 2. The algorithm maintains a memory of “good” solutions so there’s less risk of the algorithm moving in a bad direction and getting stuck there 3. It’s shown great performance in a wide variety of domains
The performance improvement with PSO included was immediately obvious.
The last thing that was needed was a name. GPU-Accelerated LocaLOptimisation and Particle swarm provides both a description of the algorithm and an acronym that gives a hat-tip to DASH. Perfect!
The GALLOP algorithm
As described above, GALLOP is a hybrid algorithm which contains two components, a local optimiser and a particle swarm optimiser. These are described in more detail below.
Local optimisation
The local optimisation algorithm used in GALLOP by default is Adam. This algorithm is very popular for training neural networks, and efficient implementations are available in almost every deep learning library.
Adam incorporates two distinct innovations that improve its performance relative to (stochastic) gradient descent.
Adam has a per-parameter adaptive step size in addition to a single global step size used for all parameters. This is useful as different degrees of freedom will have different effects on \(\chi^2\) for the same percentage change in the parameter value. For example, the translation of a whole molecule within a unit cell affects the position of more scattering atoms than changing a torsion angle. What’s nice is that Adam automatically adjusts the step size for each parameter as it goes, meaning that a suitable step size is used throughout optimisation.
Adam incorporates momentum, which helps it to escape shallow local minima, pass rapidly through flat regions of the hypersurface and dampens uncesessary oscillations in the optimisation trajectory. For an excellent overview of momentum (with a focus on ML applications), see this article: https://distill.pub/2017/momentum/
Adam
Using the gradient obtained by automatic differentiation, \(\textbf{g}_t\), Adam stores exponentially decaying averages of the gradients, \(\textbf{m}_t\), and squared gradients, \(\textbf{v}_{t}\), which are then used in conjunction with the overall step size, \(\alpha\), to give a suitable step size for each parameter being optimised. The parameters \(\beta_1\) and \(\beta_2\) are numbers less than one that control the rate at which the past gradients and squared gradients respectively decay.
Because \(\textbf{m}_t\) and \(\textbf{v}_{t}\) are initialised as vectors of zeros, the authors of Adam use the following corrective terms to reduce the effect of this biasing, which can be particularly problematic in the early stages of optimisation:
These bias corrected terms are then used to update the parameters to be optimised, \(\textbf{x}_t\), where \(\epsilon\) is included to prevent numerical errors:
The authors of Adam suggest default parameters of \(\beta_1 = 0.9\), \(\beta_2 = 0.999\) and \(\epsilon = 1 \times 10^{-8}\). The step size, \(\alpha\), must be set by the user. By default GALLOP sets \(\beta_2 = 0.9\) which decays the past squared gradients more rapidly, and was found in our testing to be more effective than the default value.
Learning rate finder
To make life easy for end users (and myself), I wanted a way to avoid having to experiment to find a suitable step size (\(\alpha\)) to use in GALLOP.
The deep learning library, fast.ai includes a heuristic known as the learning rate finder, which is used to set the step size (referred to as the learning rate in ML-parlance) for deep learning experiments automatically. This is used in conjunction with a step-size alteration policy which is carried out during optimisation, as described here.
After some testing and experimentation, GALLOP now makes use of a slightly modified version, as described below.
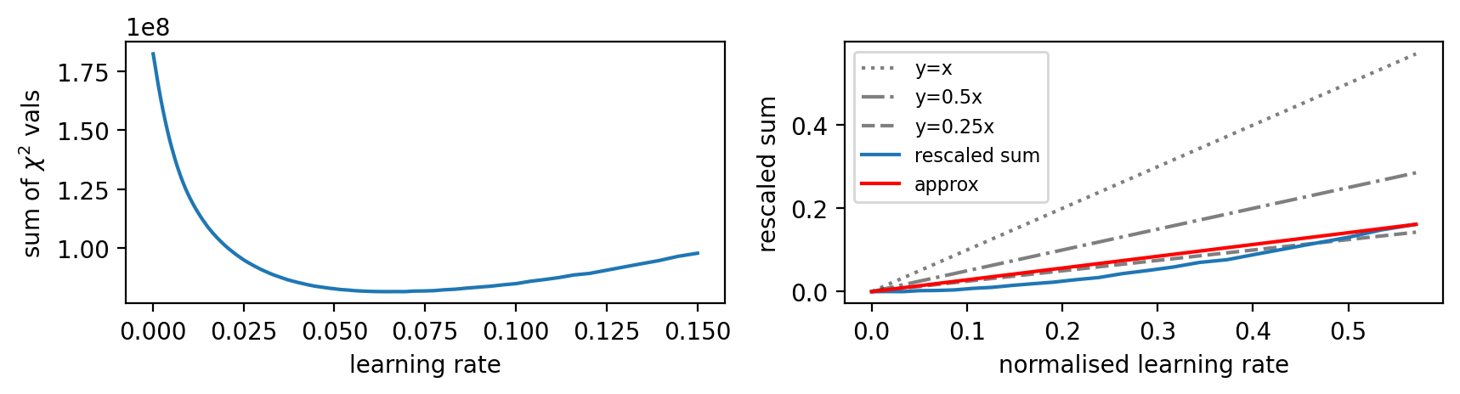
A set of 200 log-linearly spaced learning rates are initialised, ranging from \(1 \times 10^{-4}\) and \(0.15\). Starting with the smallest, GALLOP is run on the structure of interest, and the step size increased to the next value after every iteration. The sum of the \(\chi^2\) values is recorded after each iteration, and subsequently plotted.
The minimum point on this plot, \(\alpha_{min}\), is then used to give the step size. It may be scaled after considering the gradient of the line as the step size is increased beyond \(\alpha_{min}\).
To do this, the step sizes and \(\chi^2\) values are rescaled into the range 0-1. The x-axis is shifted such that \(\alpha_{min}\) sits at 0, and the data plotted. If the gradient of the resultant curve (approximated by the red straight line below) is > 0.5, then this is considered steep. A steep gradient implies a high sensitivity to the step size, and hence \(\alpha_{min}\) is scaled by a factor of 0.5, i.e. GALLOP runs with a step size of \(0.5\alpha_{min}\). A medium gradient (between 0.25 and 0.5) results in multiplication factor of 0.75, whilst a shallow gradient (less than 0.25) implies relative insensitivity to the step size, and hence results in a multiplication factor of 1.0.
In the GALLOP browser interface, this information is provided in a plot:
Step size finder
In my testing, the learning rates obtained in this manner provide a good first attempt for GALLOP and provide reasonable performance over a wide variety of structures. However, this doesn’t mean that they are optimal, and if a structure isn’t solving, it might be worth looking at this parameter more closely. I tend to find that a learning rate of 0.03 - 0.05 tends to work well as a first attempt for most structures.
Particle Swarm optimisation
Particle Swarm Optimisation has previously been used in the context of SDPD in the program PeckCryst. The algorithm used in GALLOP is different to that used in PeckCryst in a number of ways which I’ll try to highlight below.
The equations for the PSO are simple. The velocity of a particle at step \(t+1\)\((\textbf{v}_{t+1})\), is calculated from the velocity at the previous step \((\textbf{v}_{t})\) and the position of the particle \((\textbf{x}_t)\) using the following equation:
Where \(\omega_{t}\) is the inertia of the particle (which controls how much the previous velocity influences the next velocity) and \(c_1\) and \(c_2\) control the maximum step size in the direction of the best particle in the swarm \((\textbf{g}_{best})\) and best position previously visited by the particle \((\textbf{x}_{best})\) respectively. In contrast to PeckCryst which uses scalars, in GALLOP, by default \(\textbf{R}_1\) and \(\textbf{R}_2\) are diagonal matrices with their elements drawn independently from a standard uniform distribution. This provides more variability in how the particles move which helps to improve the exploration. In addition, the maximum absolute velocity in each direction in GALLOP is limited to 1.0.
GALLOP calculates \(\omega\) for particle \(i\) by ranking all of the \(N\) particles in the swarm in terms of their \(\chi^2\) value, and the calculating their inertia using the following equation:
\[ \omega_i = 0.4 + \frac{Rank_i}{2N} \]
where \(Rank_i\) is the position of particle \(i\) in a sorted list of their respective \(\chi^2\) values. This gives inertia values in the range 0.4 - 0.9, and means that the best particles slow down, whilst the worst particles in the swarm have higher inertias and hence are able to continue moving more rapidly towards (hopefully) promising areas of the hypersurface.
The degrees of freedom are then updated using the velocity and previous parameters according to:
Another difference to PeckCryst is the coordinate transform that is performed in GALLOP. The fractional coordinates are transformed to account for the repeating unit cell and the fact that coordinates of -0.1 and 0.9 are equivalent. The torsion angles are also transformed to ensure that the PSO treats angles of +180 and -180 degrees as equivalent. Molecular orientations, represented in GALLOP with quaternions, do not require any transformation. For local optimisation, the degrees of freedom that are optimised are:
Following the PSO update, these are transformed back for use in LO using the two-argument arctangent function which gives values in the range \(-\pi\) to \(\pi\), which therefore necessitates additional scaling by a factor of \(1/2\pi\) for the positions.
GALLOP
Bringing it all together, this flow chart shows how the GALLOP algorithm operates:
Gallop flow chart
Typically, 500 LO steps are performed prior to a single PSO step.
The results reported here demonstrate a significant improvement in performance relative to DASH. The success rate is >30 times that of DASH, and the GPU-acceleration means that the time taken to process the runs is also significantly lower than can be accomplished without distributed computing for the DASH jobs.
In my next post, I’ll go over how to use GALLOP to solve crystal structures.